Optimize Your NWChem Workflow: Finding the Global Minimum
Finding the global minimum energy structure for a molecule is a crucial step in many computational chemistry studies. It's the foundation for accurate predictions of molecular properties, reaction mechanisms, and spectroscopic data. However, the task isn't always straightforward. The potential energy surface (PES) is often complex, riddled with local minima that can trap optimization algorithms. This article will explore strategies to optimize your NWChem workflow to efficiently locate the global minimum energy structure, even for challenging systems.
What is the Global Minimum?
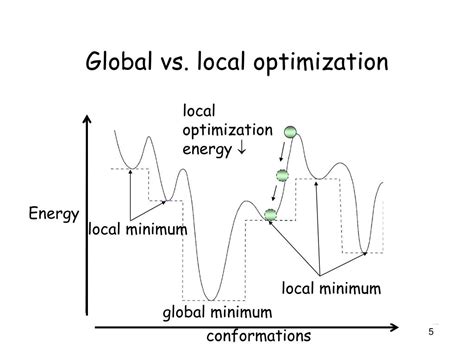
Before delving into optimization strategies, it's vital to understand the concept of the global minimum. In the context of computational chemistry, it represents the lowest energy structure a molecule can adopt. All other structures, even those with relatively low energy, are considered local minima – stable points surrounded by higher-energy configurations, but not necessarily the absolute lowest in energy. Mistaking a local minimum for the global minimum can lead to significant inaccuracies in subsequent calculations.
Common Challenges in Finding the Global Minimum
The search for the global minimum is computationally expensive and often presents several challenges:
- High dimensionality of the PES: The number of degrees of freedom increases rapidly with molecular size, making the PES incredibly complex to navigate.
- Multiple local minima: The presence of many local minima traps optimization algorithms, hindering the search for the global minimum.
- Computational cost: Exploring the entire PES exhaustively is often infeasible due to the immense computational resources required.
NWChem Optimization Methods and Strategies
NWChem offers a variety of optimization methods. The choice of method greatly influences the efficiency and success of finding the global minimum. Here are some effective strategies:
1. Choosing the Right Optimization Algorithm
NWChem provides several gradient-based optimization algorithms, each with strengths and weaknesses. For finding the global minimum, consider these:
- BFGS (Broyden–Fletcher–Goldfarb–Shanno): A quasi-Newton method that's generally efficient and robust for locating local minima. It's often a good starting point.
- L-BFGS (Limited-memory BFGS): A memory-efficient variant of BFGS, suitable for larger systems.
- Conjugate Gradient: Another effective algorithm, particularly useful when the Hessian matrix is unavailable or expensive to calculate.
Often, the best approach is a hybrid strategy. Start with a less computationally demanding method to reach a reasonable initial guess, then switch to a more sophisticated algorithm for finer refinement.
2. Multiple Starting Geometries
One of the most powerful techniques to improve your chances of finding the global minimum is to perform multiple optimizations from different initial geometries. Randomly generating several starting structures ensures you sample a wider region of the PES. This significantly increases the likelihood of escaping local minima and converging to the global minimum.
3. Simulated Annealing
Simulated annealing is a powerful stochastic method that allows the optimization algorithm to escape from local minima by accepting higher-energy configurations with a certain probability, gradually decreasing this probability over time. This method is especially effective for complex PESs with many local minima. NWChem's implementation provides a powerful tool for such searches.
4. Basin Hopping
Basin hopping is another global optimization technique that combines local optimization with random perturbations of the molecular geometry. It effectively explores different regions of the PES, increasing the chances of finding the global minimum.
5. Utilizing Different Basis Sets and Levels of Theory
The choice of basis set and level of theory can impact the accuracy of the energy calculations and consequently the identification of the global minimum. A higher level of theory generally improves accuracy but increases computational cost. A pragmatic approach is to initially use a less computationally demanding method for exploring the PES and then refine promising structures with higher-level calculations.
Troubleshooting and Common Issues
Even with the strategies outlined above, finding the global minimum can be challenging. Common issues include:
- Convergence to local minima: This is often the biggest hurdle. Employing multiple starting geometries, simulated annealing, or basin hopping are key to overcoming this.
- Slow convergence: This can be due to a poor choice of optimization algorithm or a highly complex PES. Experimenting with different algorithms and parameters is necessary.
- Numerical instability: Ensure proper convergence criteria and consider using tighter tolerances if necessary.
Conclusion
Locating the global minimum energy structure is a crucial aspect of computational chemistry. Optimizing your NWChem workflow involves careful consideration of the optimization algorithm, multiple starting geometries, and potentially global optimization techniques like simulated annealing or basin hopping. By systematically applying these strategies and addressing potential issues, you can significantly enhance your chances of successfully identifying the true global minimum energy structure for your molecule of interest. Remember that patience and experimentation are essential components of a successful computational chemistry workflow.

